[CAS NO. 112885-41-3] Mosapride
Please click "REQUEST A QUOTE" button if you need other sizes or custom synthesis
request a quote
If there is no stock, or you need other sizes or custom synthesis, please:
PRODUCTS SPECIFICATIONS [112885-41-3]
Store
Catalog
SLK-S4839
Brand
Selleck
CAS
112885-41-3
DESCRIPTION [112885-41-3]
Overview
| MDL | MFCD00867430 |
|---|---|
| Molecular Weight | 421.89 |
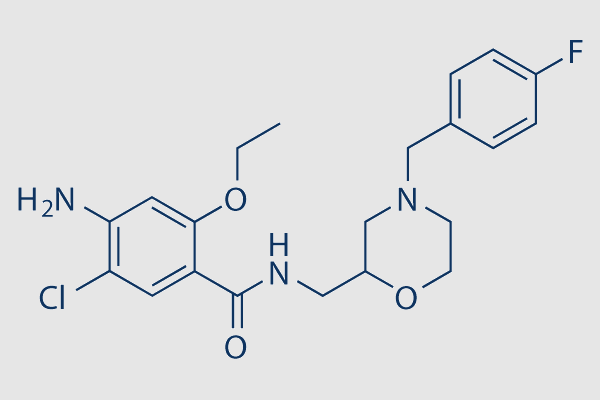
| Molecular Formula | C21H25ClFN3O3 |
| SMILES | C(NCC1CN(CC2=CC=C(F)C=C2)CCO1)(=O)C3=C(OCC)C=C(N)C(Cl)=C3 |
For research use only.
Storage
3 years,-20°C,powder
1 years,-80°C,in solvent
1 years,-80°C,in solvent
Shipping
Room temperature shipping(Stability testing shows this product can be shipped without any cooling measures.)
Preparing Stock Solutions
| 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3703 mL | 11.8514 mL | 23.7029 mL |
| 5 mM | 0.4741 mL | 2.3703 mL | 4.7406 mL |
| 10 mM | 0.2370 mL | 1.1851 mL | 2.3703 mL |
| 50 mM | 0.0474 mL | 0.2370 mL | 0.4741 mL |
Description
Mosapride is a gastroprokinetic agent that acts as a selective agonist.
Targets
| 5HT4 [1] |
In vitro
Mosapride facilitates acetylcholine release from the enteric cholinergic neurons. It does not block K+ channels or D2 dopaminergic receptors. Mosapride is a selective 5-HT4 receptor agonist with no affinity for 5-HT1, 5-HT2, adrenalineα1, adrenalineα2 or dopamine D2 receptors.
In vivo
Mosapride augmented lower gastrointestinal motility in animal models and in patients with lower gastrointestinal disorders. It enhanced defecation responses in animal models. In human, after oral administration of single doses of mosapride 5-40 mg, peak mosapride concentrations (Cmax) were reached after about 1 hour. Both the Cmax and area under the concentration-time curve from time zero to infinity (AUC∞) increased in a dose-proportional manner. There were no significant differences in the pharmacokinetic profiles of mosapride administered as single or multiple doses. Mosapride is excreted in the urine and faeces. It is primarily metabolized in the liver by cytochrome P450 3A4 to the active metabolite, a des-4-fluorobenzyl compound.
References
Synonyms
Benzamide, 4-amino-5-chloro-2-ethoxy-N-[[4-[(4-fluorophenyl)methyl]-2-morpholinyl]methyl]-
4-Amino-5-chloro-2-ethoxy-N-[[4-[(4-fluorophenyl)methyl]-2-morpholinyl]methyl]benzamide
Mosapride
Moza
4-Amino-5-chloro-2-ethoxy-N-[[4-(4-fluorobenzyl)-2-morpholinyl]methyl]benzamide
Powered by Arctom Copyright © 2026 Arctom. All rights
reserved.