[CAS NO. 1629268-00-3] ARS-853 (ARS853)
Please click "REQUEST A QUOTE" button if you need other sizes or custom synthesis
request a quote
If there is no stock, or you need other sizes or custom synthesis, please:
PRODUCTS SPECIFICATIONS [1629268-00-3]
Store
Catalog
SLK-S8156
Brand
Selleck
CAS
1629268-00-3
DESCRIPTION [1629268-00-3]
Overview
| MDL | MFCD30532658 |
|---|---|
| Molecular Weight | 432.94 |
| Molecular Formula | C22H29ClN4O3 |
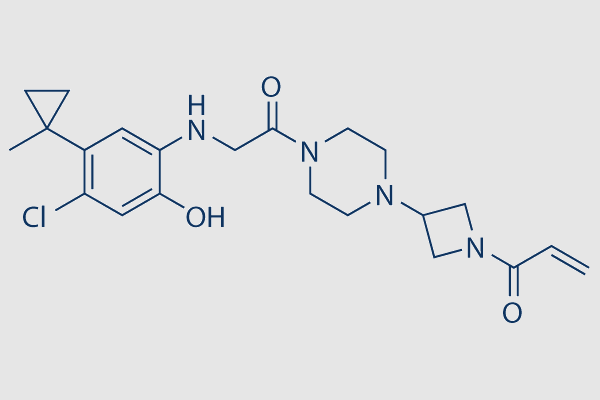
| SMILES | C=CC(N1CC(N2CCN(C(CNC3=CC(C4(C)CC4)=C(Cl)C=C3O)=O)CC2)C1)=O |
For research use only.
Storage
3 years,-20°C,powder
1 years,-80°C,in solvent
1 years,-80°C,in solvent
Shipping
Room temperature shipping(Stability testing shows this product can be shipped without any cooling measures.)
Description
ARS-853 is a selective, covalent inhibitor that inhibits mutant KRAS-driven signaling by binding to the GDP-bound oncoprotein and preventing activation. ARS-853 also induces .
Targets
| K-Ras(G12C) [2] (in H358 cells) |
| 2.5 μM |
In vitro
ARS-853 treatment of KRAS cells led to a dose-dependent and nearly complete inhibition of CRAF-RBD (RBD)-mediated pulldown of KRAS from lysates, with an IC50 of approximately 1 μmol/L. Treatment of H358 cells by ARS-853 resulted in a significant loss of KRAS–CRAF interactions. Consistent with an inactive state of KRAS once bound to ARS-853, downstream signaling through both MAPK (including pMEK, pERK, and pRSK) and PI3K signaling (pAKT) pathways was inhibited by ARS-853 in H358 and other KRAS cell lines. The inhibition of RAF-RBD pulldown and KRAS downstream signaling was sustained over a period of 72 hours, accompanied by G1 cell-cycle arrest, loss of Cyclin D1 and Rb expression, and an increase in the cell-cycle inhibitor p27 KIP1. In addition, hallmarks of apoptosis, including cleaved PARP and increases in sub-diploid DNA, were observed in H358 cells following treatment with ARS-853. No effects on RAF-RBD binding or downstream signaling were observed in A549 cells (KRASG12S), and the inhibitory effects of ARS-853 in H358 cells could be rescued by ectopic expression of KRAS. KRAS is the most potent covalent target of ARS-853 across more than 2,700 cellular proteins and consistently find that this compound exerts no effects on cellular signaling or growth in non-KRAS cells at concentrations up to 10-fold higher than its KRAS potency. ARS-853 reacts only with the inactive (GDP-bound), but the not the active (GTP-bound), state of KRAS. ARS853 reduced KRAS-GTP levels and ERK phosphorylation in human embryonic kidney 293 (HEK293) or H358 cells engineered to express KRAS but not in those expressing KRAS. ARS853 traps KRAS in a GDP-bound conformation by lowering its affinity for nucleotide exchange factors.
References
Powered by Arctom Copyright © 2026 Arctom. All rights
reserved.