[CAS NO. 1628838-42-5] RAF709
Please click "REQUEST A QUOTE" button if you need other sizes or custom synthesis
request a quote
If there is no stock, or you need other sizes or custom synthesis, please:
PRODUCTS SPECIFICATIONS [1628838-42-5]
Store
Catalog
SLK-S8690
Brand
Selleck
CAS
1628838-42-5
DESCRIPTION [1628838-42-5]
Overview
| MDL | MFCD30534393 |
|---|---|
| Molecular Weight | 542.55 |
| Molecular Formula | C28H29F3N4O4 |
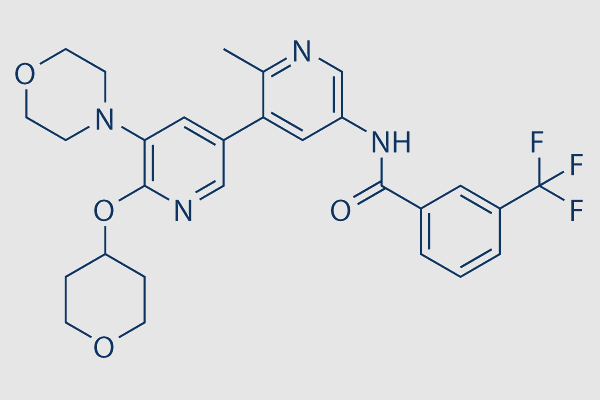
| SMILES | O=C(NC1=CN=C(C)C(C2=CC(N3CCOCC3)=C(OC4CCOCC4)N=C2)=C1)C5=CC=CC(C(F)(F)F)=C5 |
For research use only.
Storage
3 years,-20°C,powder
1 years,-80°C,in solvent
1 years,-80°C,in solvent
Shipping
Room temperature shipping(Stability testing shows this product can be shipped without any cooling measures.)
Preparing Stock Solutions
| 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.8431 mL | 9.2157 mL | 18.4315 mL |
| 5 mM | 0.3686 mL | 1.8431 mL | 3.6863 mL |
| 10 mM | 0.1843 mL | 0.9216 mL | 1.8431 mL |
| 50 mM | 0.0369 mL | 0.1843 mL | 0.3686 mL |
Description
RAF709 is a potent inhibitor of kinase with almost equivalent IC50 values of 0.4 nM for B-RAF and C-RAF, showing a high level of selectivity, demonstrating greater than 99% on-target binding to BRAF, BRAFV600E, and CRAF at 1 μM and very few off-targets with DDR1 (>99%), DDR2 (86%), FRK (92%), and PDGFRb (96%), the only kinases with binding >80% at 1 μM.
Targets
In vitro
RAF709 appears to have very slow dissociation kinetics (T > 6.5 h) using the rapid dilution method to measure its dissociation rate constant. In cellular assays, the dose−response of pMEK and pERK are measured in Calu-6 cells with EC50 of 0.02 and 0.1 μM with minimal paradoxical activation and inhibition of proliferation with EC50 of 0.95 μM. RAF709 stabilizes BRAF−CRAF dimers with an EC50 of 0.8 μM. Of the 456 kinases tested, RAF709 shows a high level of selectivity, demonstrating greater than 99% on-target binding to BRAF, BRAFV600E, and CRAF at 1 μM and very few off-targets with DDR1 (>99%), DDR2 (86%), FRK (92%), and PDGFRb (96%), the only kinases with binding >80% at 1 μM. RAF709 shows equal activity against both RAF monomers and dimers. In in vitro biochemical assays, RAF709 exhibits potent inhibitory activity targeting BRAF, BRAFV600E, and CRAF with IC50 values ranging between 0.3 to 1.5 nmol/L. RAF709 treatment leads to a dose-dependent induction of B/CRAF heterodimerization in HCT116, but inhibits MEK and ERK phosphorylation, in line with the ability of RAF709 to effectively inhibit the RAF dimers. RAF709 selectively inhibits oncogenic signaling and proliferation in tumor cells with BRAF, NRAS, or KRAS mutations with minimal paradoxical activation.
In vivo
RAF709 is well tolerated and efficacious in KRAS mutant xenograft models. It is reasonably stable in plasma after a 3 h incubation at 37℃ across species [plasma stability (%remaining): rat 85%, mouse 82%, dog 95%, human 101%], and plasma protein binding is measured to be 98% across species. In pharmacokinetic experiments, RAF709 has moderate clearance in mouse (35 mL/min/kg) and dog (14 mL/min/kg) and high clearance in rat (50 mL/min/kg). Cmax in mouse (1 μM), dog (0.5 μM), and rat (0.5 μM) reach pharmacologically active concentrations, and acceptable oral availability is observed in mouse (68%), rat (24%), and dog (48%). In the Calu-6 xenograft nude mouse model, treatment with RAF709 results in dose-dependent antitumor activity with 10 mg/kg being subefficacious (%T/C = 92%), 30 mg/kg resulted in measurable antitumor activity (% T/C = 46%), and 200 mg/kg resulted in mean tumor regression of 92%, while the same high dose is not efficacious in the PC3, KRAS WT mode.
References
Powered by Arctom Copyright © 2026 Arctom. All rights
reserved.